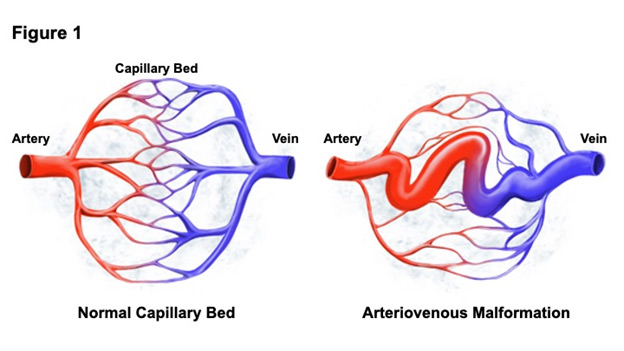
The Beleford Lab studies underlying changes in molecular signaling that cause vascular malformations (abnormal development of blood vessels) in Hereditary Hemorrhagic Telangiectasia and other genetic vascular conditions. Hereditary Hemorrhagic Telangiectasia (HHT) is a rare Mendelian vascular condition that is characterized by arteriovenous malformations (AVMs), direct connections between arteries and veins without intervening capillary beds (Figure 1). AVMs may form in any organ but are most often detected in the lung, liver, brain, and GI tract and confer significant mortality risk from devastating rupture or hemorrhage. HHT derives its name from small, dilated blood vessels (telangiectases) that form in the skin and mucosal surfaces of most patients. HHT is caused by heterozygous loss of function of ENG or ACVRL1 (aka ALK1), genes that encode endothelial receptors for TGFβs and BMPs. SMAD4, the common co-SMAD for TGFβ and BMP9 signaling, causes 1% of HHT cases. A fundamental question to the study of HHT pathobiology is why some patients develop vascular malformations while others do not. We hypothesize that the genetic modifier PTPN14 (protein tyrosine phosphatase, nonreceptor, 14) modifies and potentiates vascular malformations in HHT and other vascular conditions. It has previously been shown that single nucleotide polymorphisms within PTPN14 associate with pulmonary AVMs in two large HHT cohorts. Our preliminary data suggest that the PTPN14 gene product binds and stabilizes BMP9 signaling components in endothelial cells, inhibiting ubiquitin-mediated protein turnover and promoting vascular stability. The Beleford Lab seeks to clarify the molecular interplay between PTPN14 and BMP9, TGFβ, and HIPPO signaling pathway components with the goal of understanding what causes the loss of vascular integrity in HHT and other severe vascular conditions in order to ultimately tailor targeted therapeutic interventions.